Application of mendelian randomization methods using expression data
Yan Holtz, Zhihong Zhu, Julanne Frater, Perry Bartlett, Jian Yang, John McGrath
This document runs SMR on the vitamin-D GWAS summary statistics. The goal is to understand which gene is hidden behind each of the SNP detected in the GWAS. The SMR software and its documentation is available for download here. We first run SMR using the eQTLgen dataset. We then confirmed our results on the GTEx dataset in different tissue.
Method
The GWAS result suggests an oligogenic control of the VitaminD concentration. Six major loci are described, with several genes present in the corresponding regions. We can run an SMR analysis to go deeper and find out what genes are probably involved in the control of VitaminD.
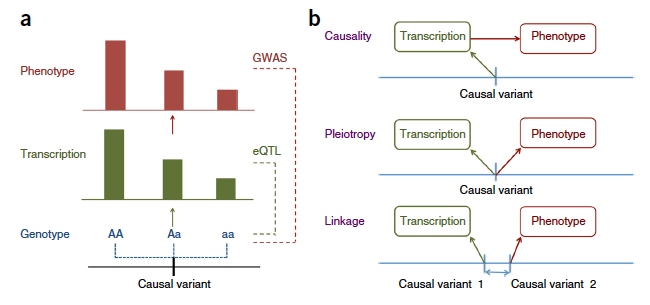
Figure: Association between gene expression and phenotype trough genotypes. (a) A model of causality where a difference in phenotype is caused by a diffference in genotype mediated by gene expression (transcription). (b) Three possible explanations for an observed association between a trait and gene expression through genotypes. From Zhihong Zhu et al.
If an association between a gene and VitaminD is detected (figure a), several options exist (figure b):
- Causality or Pleiotropy: in this case the Heidi test will be significant
- Linkage: in this case the Heidi test won’t be significant.
Data
To run SMR several datasets are required:
- bfile: individual-level SNP genotype data. I’m gonna use the HapMap3 data set:
/gpfs/gpfs01/polaris/Q0286/UKBiobank/v2EURu_HM3/ukbEURu_imp_chrin delta. - gwas-summary: the summary statistic of the vitamin-D GWAS of Xia et al. Must be in the ‘ma’ format. File will be:
/shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/1_GWAS/GWAS_vitaminD_XiaEtAL.ma - beqtl-summary: eQTL summary data. Describes the link between SNPs and gene expression.
We used the 32K eQTLGen cis-eQTL dataset as discovery dataset. Not published yet. This dataset has been provided by Annique after consurtium authorization. It is abvailable on Delta here: /shares/compbio/Group-Wray/YanHoltz/DATA/EQTL/eQTL_DATA_EQTLGEN_CONSORTIUM_32K.
We then used the GTExV7 eQTL dataset as a validation dataset. Several tissues were considered: Liver, Blood, and Brain. These datasets are also available on Delat: /gpfs/gpfs01/polaris/Q0286/GTExV7/Summary/besd/
A note on the eQTL format:
- the
.epifile gives the list of probe involved, with name, chromosome, position, associated gene. - the
.esifile gives the list of all the involved SNP, with name, chromosome, position and allele. - the
.besdis compressed and display all the association between SNP and probe.
A note on the GTEx dataset: very well described here.
- Liver: 175 samples
- Kidney: 45 samples
- Whole Blood: 407 samples
- Brain: several different tissues with sample size between 88 and 173
Discovery eQTL dataset: description
The discovery eQTL dataset is the eQTLGen dataset. Gene names are provided using Ensembl IDs, so I use the biomaRt library to get the real gene names:
# Load SMR result
eqtlgen <- read.table("0_DATA/smr_VitaminDXiaEtAl_eQTLGEN.smr", header=T)
# Find out the genes IDs (ensemble IDs are provided with this dataset)
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
tmp <- getBM(filters= "ensembl_gene_id", attributes= c("ensembl_gene_id", "hgnc_symbol", "description"), values=eqtlgen$Gene, mart= mart)
eqtlgen <- merge( eqtlgen, tmp, by.x="Gene", by.y="ensembl_gene_id", all.x=TRUE)15127 eQTLs are provided: association between probe and gene expression. This concerns 15127 unique probes, 15127 unique genes and 13713 SNPs spread in 22 chromosomes.
Can we have a look to the existing eQTL in the GWAS hit regions?
GC | chr4
There are 5 eQTL in the region of the GC gene = on chromosome 4 between 71 and 74 Mb. However the GC gene itself is NOT involved. The gene SLC4A4 nearby is controled by the SNP rs12330929 that is NOT in our vitaminD hit area. We have NO chance to detect GC as associated in this SMR analysis.
eqtlgen %>%
filter(topSNP_chr==4) %>%
filter(topSNP_bp>71000000 & topSNP_bp<74000000) %>%
dplyr::select(hgnc_symbol, description, ProbeChr, Probe_bp, topSNP, topSNP_bp, A1, A2, Freq, p_eQTL)CYP2R1 | chr11 | ~15 Mb
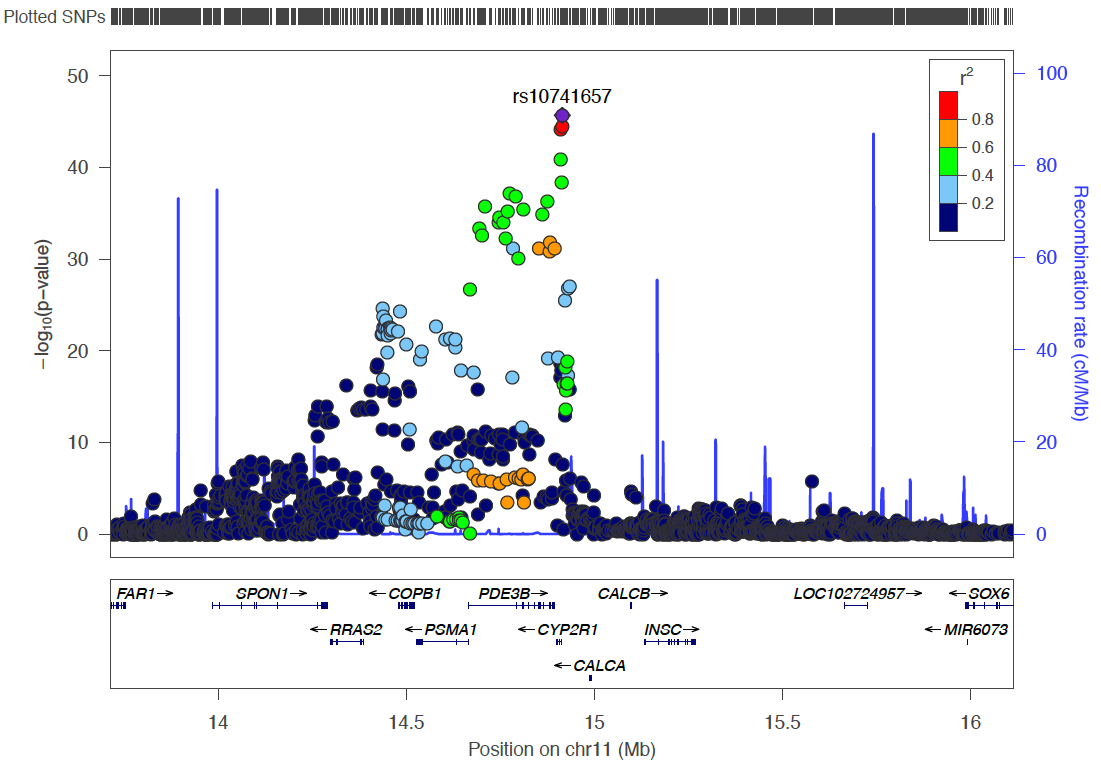
There are 5 eQTL in the region of the CYP2R1 gene = Between 14 and 15.5 Mb on chromosome 11. They concern the genes RRAS2, COPB1, PDE3B, CYP2R1 and INSC. CYP2R1 is our candidate gene since the top hit SNP is in this gene
eqtlgen %>%
filter(topSNP_chr==11) %>%
filter(topSNP_bp>14000000 & topSNP_bp<15500000) %>%
dplyr::select(hgnc_symbol, description, ProbeChr, Probe_bp, topSNP, topSNP_bp, A1, A2, Freq, p_eQTL)NADSYN1 / DHCR7 | chr11 | ~71 Mb
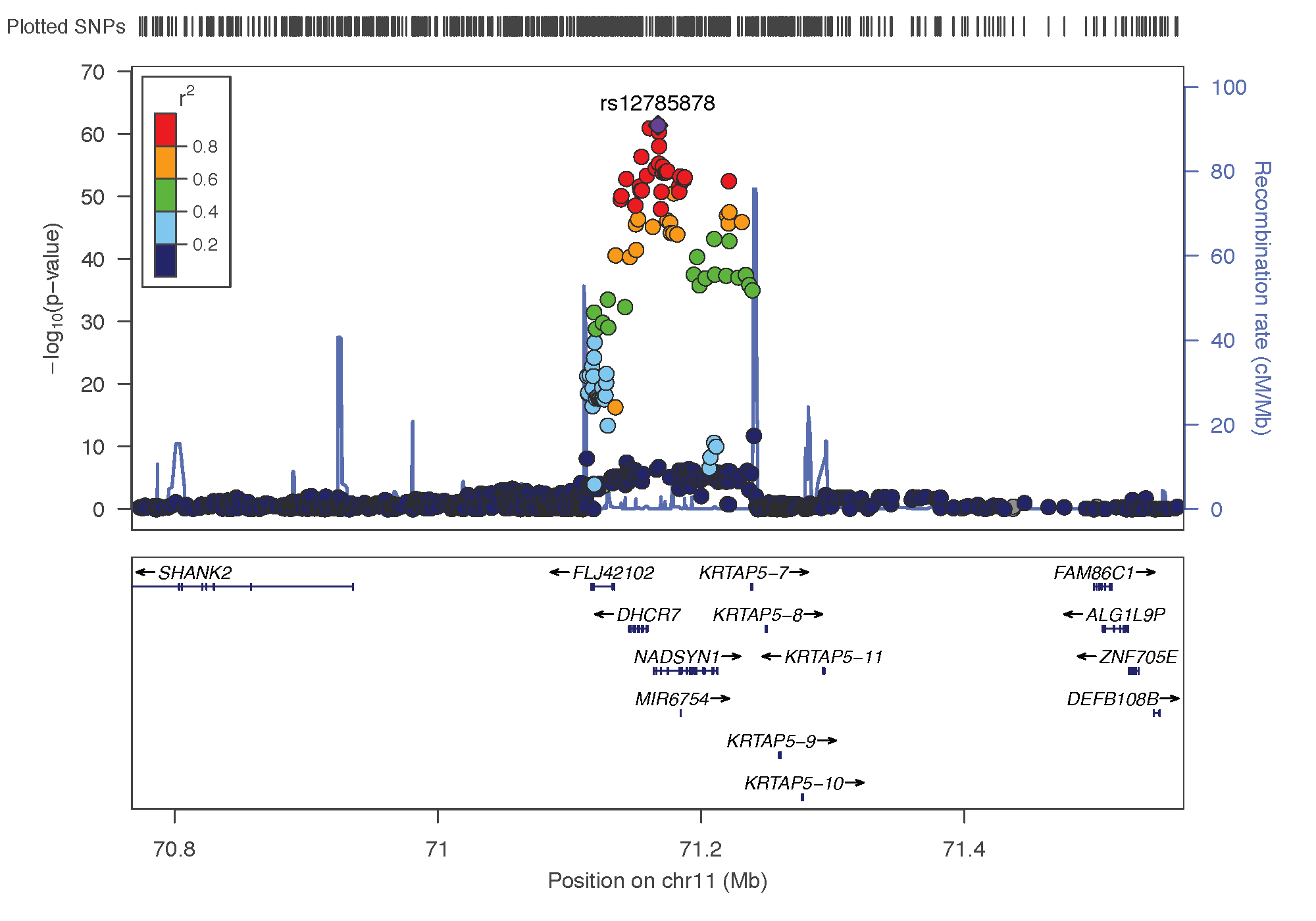
There are 4 eQTL in the region of the DHCR7 gene = Between 70 and 72 Mb on chromosome 11. Genes concerned are DHCR7, AP002387.1, NADSYN1, FAM86C1. Our target are here.
# One gene is missing in biomaRt
eqtlgen[which(eqtlgen$Gene=="ENSG00000254682") , "hgnc_symbol"] <- "AP002387.2"
eqtlgen %>%
filter(topSNP_chr==11) %>%
filter(topSNP_bp>70800000 & topSNP_bp<71500000) %>%
dplyr::select(hgnc_symbol, description, ProbeChr, Probe_bp, topSNP, topSNP_bp, A1, A2, Freq, p_eQTL)- Keep in mind how the chromosome 11 looks like
Chromosome 11 ~ 15Mb
Chromosome 11 ~ 71Mb
Run SMR on discovery dataset
The first step of SMR consists to run the algorythm on every significant eQTL (eQTL = significant relationship between a SNP allele and the expression of a gene). For each gene involved in an eQTL, we are going to test its putative effect on our trait (vitaminD) thanks to the GWAS summary statistic of this trait.
This work is done on the eQTLGen 32k dataset:
# Good directory
cd /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K
# Run the analysis
tmp_command="smr_Linux --bfile /gpfs/gpfs01/polaris/Q0286/UKBiobank/v2EURu_HM3/ukbEURu_imp_chr{TASK_ID}_v2_HM3_QC --gwas-summary /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/1_GWAS/GWAS_vitaminD_XiaEtAL.ma --beqtl-summary /shares/compbio/Group-Wray/YanHoltz/DATA/EQTL/eQTL_DATA_EQTLGEN_CONSORTIUM_32K/cis-eQTLs-full_formatted.txt_besd-dense --out smr_VitaminDXiaEtAl_{TASK_ID} --thread-num 1"
qsubshcom "$tmp_command" 1 30G smr_VitaminD 10:00:00 "-array=1-22"
# Concatenate chromosome results
cat smr_VitaminDXiaEtAl_*smr | head -1 > smr_VitaminDXiaEtAl.smr
cat smr_VitaminDXiaEtAl_*smr | grep -v "^probeID" >> smr_VitaminDXiaEtAl.smr
mv smr_VitaminDXiaEtAl.smr smr_VitaminDXiaEtAl_eQTLGEN.smr
# Transfer locally
cd /Users/y.holtz/Dropbox/QBI/4_UK_BIOBANK_GWAS_PROJECT/VitaminD-GWAS/0_DATA
scp y.holtz@delta.imb.uq.edu.au:/shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K/smr_VitaminDXiaEtAl_eQTLGEN.smr .Result
The SMR threshold used in our study takes into account multiple testing. It is thus 0.05 / # of test. 6 have been detected by SMR. Here is their detail:
thres <- (0.05 / nrow(eqtlgen))
eqtlgen %>% filter(p_SMR < thres ) %>% arrange(ProbeChr, Probe_bp)Three associations are located at 2 regions detected in the GWAS analysis.
- on chromosome 11 ~ 15Mb,
- on chromosome 11 ~ 71 MB.
Three models are consistent with a significant association from the SMR test using only a single genetic variant. These models are causality (Z - > X - > Y), pleiotropy (Z ->X and Z ->Y), and linkage (Z1->X, Z2->Y, and Z1 and Z2 are in LD). I used the heterogeneity in dependent instruments (HEIDI) test proposed by Zhu et al. to detect the linkage situation that is of less biological interest.
Only the gene DHCR7 passed the HEIDI test and can thus be considered as causal on VitaminD.
Here is the diagram showing the organization of these 2 SMR regions:
Locus ~ 15 Mb
# Good folder
cd /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K
# Load the position of genes:
wget https://www.cog-genomics.org/static/bin/plink/glist-hg18
# Send smr plot for this loci (first of chromosome 11)
tmp_command="smr_Linux --bfile /gpfs/gpfs01/polaris/Q0286/UKBiobank/v2EURu_HM3/ukbEURu_imp_chr11_v2_HM3_QC --gwas-summary /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/1_GWAS/GWAS_vitaminD_XiaEtAL.ma --beqtl-summary /shares/compbio/Group-Wray/YanHoltz/DATA/EQTL/eQTL_DATA_EQTLGEN_CONSORTIUM_32K/cis-eQTLs-full_formatted.txt_besd-dense --out myplot --plot --probe ENSG00000152270 --probe-wind 500 --gene-list /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K/glist-hg18"
qsubshcom "$tmp_command" 1 30G smr_plot_vitaminD_loc1 10:00:00 ""
# Send smr plot for this loci (Second of chromosome 11)
tmp_command="smr_Linux --bfile /gpfs/gpfs01/polaris/Q0286/UKBiobank/v2EURu_HM3/ukbEURu_imp_chr11_v2_HM3_QC --gwas-summary /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/1_GWAS/GWAS_vitaminD_XiaEtAL.ma --beqtl-summary /shares/compbio/Group-Wray/YanHoltz/DATA/EQTL/eQTL_DATA_EQTLGEN_CONSORTIUM_32K/cis-eQTLs-full_formatted.txt_besd-dense --out myplot --plot --probe ENSG00000254682 --probe-wind 500 --gene-list /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K/glist-hg18"
qsubshcom "$tmp_command" 1 30G smr_plot_vitaminD_loc1 10:00:00 ""
# transfer locally
cd /Users/y.holtz/Dropbox/QBI/4_UK_BIOBANK_GWAS_PROJECT/VitaminD-GWAS/0_DATA
scp y.holtz@delta.imb.uq.edu.au:/shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_EQTLGEN_32K/plot/* .# Make the plot
source("SCRIPT/plot_SMR.r")
# Read the data file in R:
SMRData = ReadSMRData("0_DATA/SMR_PLOT/myplot.ENSG00000152270.txt")
# Plot the SMR results in a genomic region centred around a probe:
SMRLocusPlot(data=SMRData, smr_thresh=8.4e-6, heidi_thresh=0.05, plotWindow=1000, max_anno_probe=16)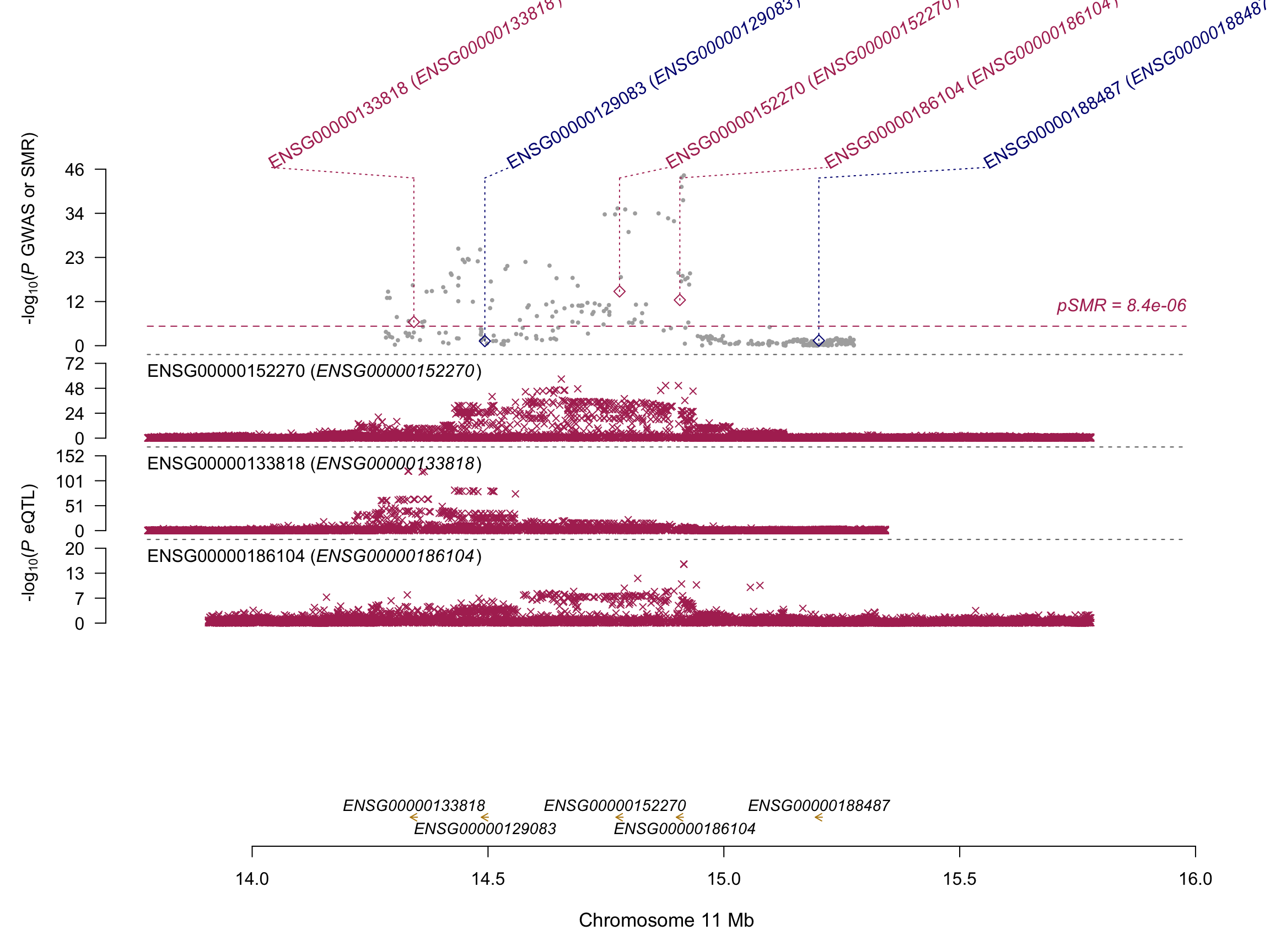
Locus ~ 71 Mb
# Read the data file in R:
SMRData = ReadSMRData("0_DATA/SMR_PLOT/myplot.ENSG00000254682.txt")
# Plot the SMR results in a genomic region centred around a probe:
SMRLocusPlot(data=SMRData, smr_thresh=8.4e-6, heidi_thresh=0.05, plotWindow=1000, max_anno_probe=16)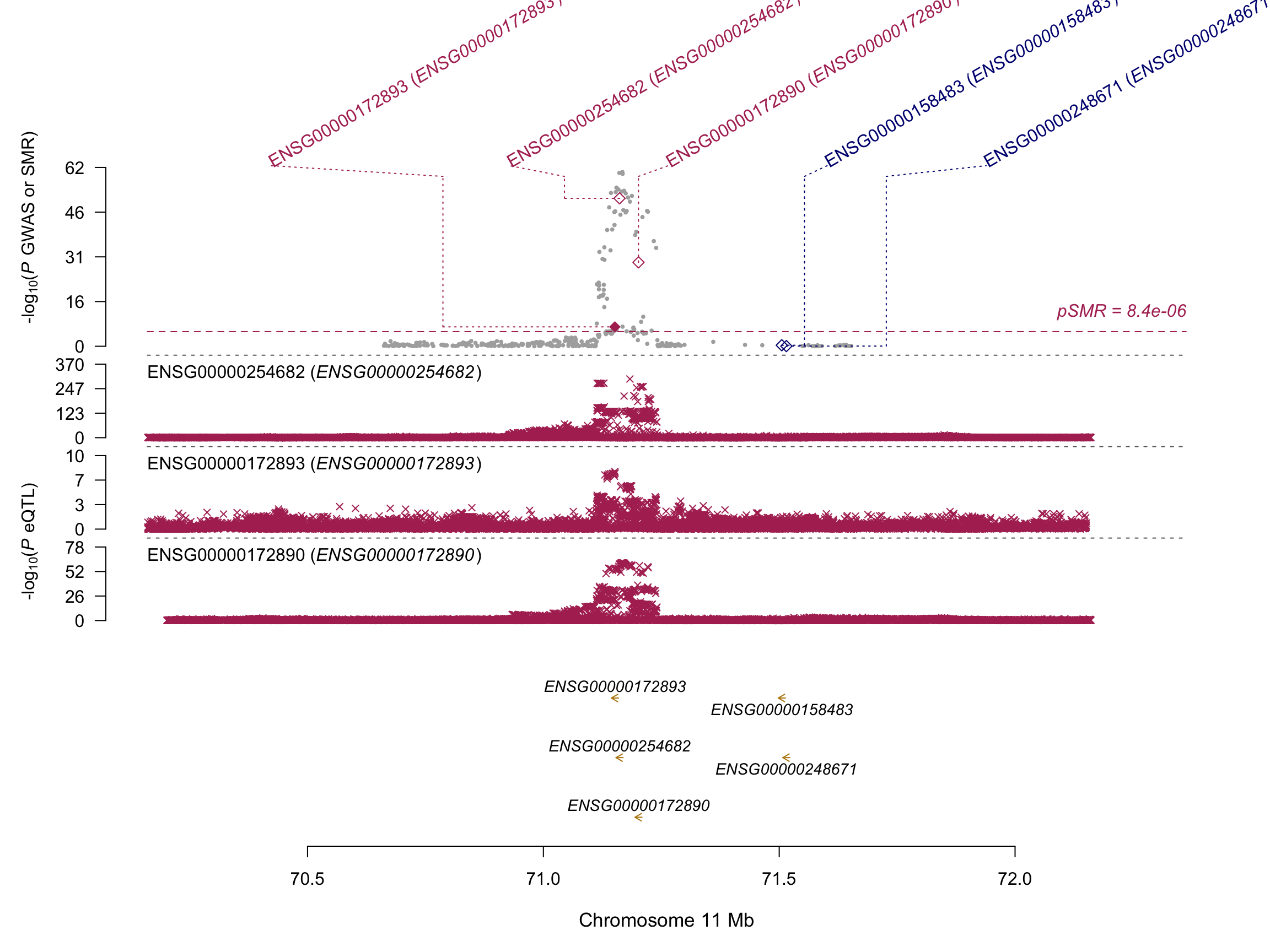
Do we have other association under pval=0.01 but that did not survive multiple testing correction in this area:
eqtlgen %>%
filter(p_SMR < 0.01) %>%
filter(p_SMR > (0.05 / nrow(eqtlgen)) ) %>%
arrange(ProbeChr, as.numeric(as.character(Probe_bp)))Run SMR on validation dataset
To replicate the SMR associations in a wider range of tissues relevant to VitaminD, we performed SNP analyses based on cis-eQTL data from several tissues in GTEx:
- Liver
- Whole blood,
- Brain (13 different tissues)
# Good directory
cd /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_GTEX
# Run the analysis on several tissues:
for i in $(echo Liver Whole_Blood Brain_Amygdala Brain_Anterior_cingulate_cortex_BA24 Brain_Caudate_basal_ganglia Brain_Cerebellar_Hemisphere Brain_Cerebellum Brain_Cortex Brain_Frontal_Cortex_BA9 Brain_Hypothalamus Brain_Hippocampus Brain_Nucleus_accumbens_basal_ganglia Brain_Putamen_basal_ganglia Brain_Spinal_cord_cervical_c-1 Brain_Substantia_nigra ) ; do
echo $i
tmp_command="smr_Linux --bfile /gpfs/gpfs01/polaris/Q0286/UKBiobank/v2EURu_HM3/ukbEURu_imp_chr{TASK_ID}_v2_HM3_QC --gwas-summary /shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/1_GWAS/GWAS_vitaminD_XiaEtAL.ma --beqtl-summary /gpfs/gpfs01/polaris/Q0286/GTExV7/Summary/besd/${i} --out smr_VitaminDXiaEtAl_GTEX${i}_{TASK_ID} --thread-num 1"
qsubshcom "$tmp_command" 1 30G smr_VitaminD 10:00:00 "-array=1-22"
done
# Concatenate chromosome results
cat smr_VitaminDXiaEtAl_GTEX*smr | head -1 | awk '{print "dataset",$0}' > res_smr_VitaminDXiaEtAl_GTEX.smr
for i in $(echo Liver Whole_Blood Brain_Amygdala Brain_Anterior_cingulate_cortex_BA24 Brain_Caudate_basal_ganglia Brain_Cerebellar_Hemisphere Brain_Cerebellum Brain_Cortex Brain_Frontal_Cortex_BA9 Brain_Hypothalamus Brain_Hippocampus Brain_Nucleus_accumbens_basal_ganglia Brain_Putamen_basal_ganglia Brain_Spinal_cord_cervical_c-1 Brain_Substantia_nigra ) ; do
echo $i
cat smr_VitaminDXiaEtAl_GTEX${i}*smr | grep -v "^probeID" | awk -v a="$i" '{print a,$0}' >> res_smr_VitaminDXiaEtAl_GTEX.smr
done
# Clean
rm smr* qsub*
# Transfer locally
cd /Users/y.holtz/Dropbox/QBI/4_UK_BIOBANK_GWAS_PROJECT/VitaminD-GWAS/0_DATA
scp y.holtz@delta.imb.uq.edu.au:/shares/compbio/Group-Wray/YanHoltz/VITAMIND_XIA_ET_AL/5_SMR/ON_GTEX/res_smr_VitaminDXiaEtAl_GTEX.smr .The following figure represent all the genes being involved in at least 1 eQTL, for the zone of interest.
- Gene names are displayed on the X axis.
- The SMR p-value is displayed on the Y axis.
- Point color denotes the eQTL tissue.
- Point shape: dot if SMR and HEIDI tests are OK, ‘+’ otherwise.
- Hover points for more info like the HEIDI p-value.
- The significant threshold is calculated on the eQTLgen dataset with multiple testing correction.
Note and observation: - It looks like AP002387.2, RP11-660L16.2 and NADSYN1 are more or less the same gene? - Brain related tissues give a strong signal for the genes KRTAP** that are keratin associated protein.
data=read.table("0_DATA/res_smr_VitaminDXiaEtAl_GTEX.smr", header=T)
# Add the eQTLGen results
eqtlgen$dataset <- "eqtlgen"
eqtlgen$Gene <- eqtlgen$hgnc_symbol
eqtlgen <- eqtlgen[ , c( 24, 1:21) ]
data <- rbind(data, eqtlgen)
# Show all the relationships
tmp <- data %>%
filter( (ProbeChr==20 & Probe_bp>50500000 & Probe_bp<54000000) | (ProbeChr==4 & Probe_bp>70500000 & Probe_bp<75000000) | (ProbeChr==11 & Probe_bp>13500000 & Probe_bp<16500000) | (ProbeChr==11 & Probe_bp>70500000 & Probe_bp<71800000) ) %>%
mutate( ProbeChr=paste("Chromosome ", ProbeChr, sep="")) %>%
mutate( ProbeChr=ifelse(ProbeChr=="Chromosome 11" & Probe_bp<16500000, "Chromosome 11 (~15Mb)", ProbeChr)) %>%
mutate( group=ifelse(dataset=="eqtlgen", "eqtlgen", ifelse( dataset=="Liver", "Liver", ifelse(dataset=="Whole_Blood", "whole blood", "brain") ))) %>%
arrange(ProbeChr, Probe_bp) %>%
mutate( Gene=factor(Gene, unique(Gene))) %>%
mutate(text = paste("Gene: ", Gene, "\n", "Tissue: ", dataset, "\n", "Position: ", Probe_bp, "\n", "SMR pvalue: ", p_SMR, "\n", "HEIDI p-value: ", p_HEIDI)) %>%
mutate(myshape = ifelse(p_SMR<thres & p_HEIDI>0.05, "a", "b"))
p <- tmp %>% ggplot( aes(x=Gene, y=-log10(p_SMR), color=group, text=text) ) +
geom_point( shape=ifelse(tmp$p_SMR<thres & tmp$p_HEIDI>0.05, 16, 43)) +
scale_color_manual( values=c("skyblue", "black", "orange", "forestgreen") ) +
theme_ipsum() +
facet_wrap(~ ProbeChr, ncol=1, scale="free_x") +
theme(
axis.text.x=element_text(angle=45, hjust=1),
panel.spacing = unit(0.1, "lines"),
panel.grid.major.y = element_blank(),
) +
geom_hline(yintercept = -log10(thres), color="orange") +
guides(shape=FALSE)
ggplotly(p, tooltip="text")Do I have a good correlation between eQTLgen and GTEx whole blood?
General
p <- data %>%
filter(dataset %in% c("Whole_Blood", "eqtlgen")) %>%
dplyr::select(Gene, dataset, p_SMR) %>%
arrange(Gene) %>%
filter(Gene!="") %>%
filter(! Gene %in% c("LINC01422","LINC02356", "LINC01481")) %>%
tidyr::spread(dataset, p_SMR) %>%
na.omit() %>%
mutate(text=paste("Gene: ", Gene, "\n", "SMR pVal in eQTLgen: ", eqtlgen, "\n", "SMR pVal in GTEx: ", Whole_Blood)) %>%
ggplot( aes(x=Whole_Blood, y=eqtlgen, text=text )) +
geom_point() +
theme_ipsum()
ggplotly(p, tooltip="text")Focus on small pval
p <- data %>%
filter(dataset %in% c("Whole_Blood", "eqtlgen")) %>%
filter(p_SMR<0.05) %>%
dplyr::select(Gene, dataset, p_SMR) %>%
arrange(Gene) %>%
filter(Gene!="") %>%
filter(! Gene %in% c("LINC01422","LINC02356", "LINC01481")) %>%
tidyr::spread(dataset, p_SMR) %>%
na.omit() %>%
mutate(text=paste("Gene: ", Gene, "\n", "SMR pVal in eQTLgen: ", eqtlgen, "\n", "SMR pVal in GTEx: ", Whole_Blood)) %>%
ggplot( aes(x=-log10(Whole_Blood), y=-log10(eqtlgen), text=text )) +
geom_point() +
theme_ipsum()
ggplotly(p, tooltip="text")Discussion
Six genetic regions where described in the GWAS result of Xia et al. as associated with Vitamin-D:
- rs3755967: chromosome 4 ~ 73Mb | gene: GC | Genome Browser | Role = Vitamin D transport.
- rs12785878: chromosome 11 ~ 79Mb | gene: NADSYN1 / DHCR7 | Genome Browser
- rs10741657: chromosome 11 ~ 15Mb | gene: CYP2R1| Genome Browser | known role = hydroxylation.
- rs10745742: chromosome 12 ~ 94Mb | gene: AMDHD1| Genome Browser
- rs8018720: chromosome 14 ~ 38Mb | gene: SEC23A| Genome Browser
- rs17216707: chromosome 20 ~ 52Mb | gene: CYP24A1| Genome Browser | Role = Catabolism.
- Some of these genes are clearly involved in the VitaminD Pathway described by Manousaki et al.
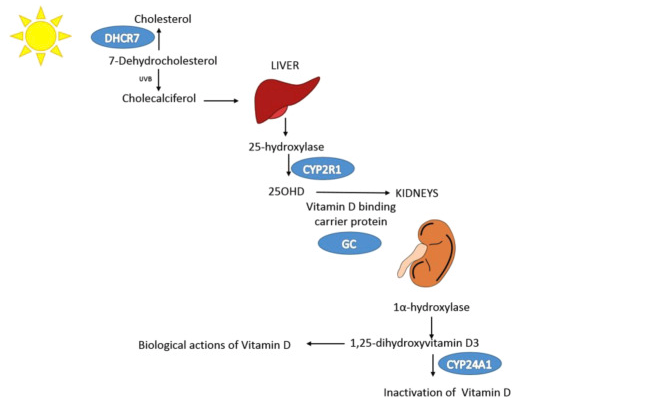
The gene GC is not involved in any eQTL in any dataset. This is surprising since it carries the SNP with the biggest effect on the GWAS. This gene is a Vitamin D binding protein. It allws to transport the vitamin D.
The gene DHCR7 is detected as causal by SMR and HEIDI tests. However, no eQTL is detected for this gene in GTEx datasets. DHCR7 is a 7-dehydrocholesterol reductase: it is involved in the cholesterol pathway.
The gene CYP2R1 is detected by SMR but does not pass the HEIDI test. This genes encodes for the cytochrome P450 2R1, which is the Vitamin D 25-hydroxylase. A low frequency SNP on this gene has a strong effect on VitaminD (paper)
The genes KRTAP* are detected as causal (both SMR and HEIDI OK) in several brain tissues. These genes are keratin associated proteins. Something to see with hair..
The gene PDE3B is detected by SMR but don’t pass the HEIDI test. It is a phosphodiesterase. It has a role in regulating heart muscle, vascular smooth muscle and platelet aggregation. This gene is not expressed in Liver, what can explain that it has not been detected using the GTEX dataset. Possible link with obesity and diabetes.
The gene RRAS2 is detected is detected by SMR but don’t pass the HEIDI test.
The gene SPON1 is detected in Liver. But nothing else to say about it…
Supplementary Material: Whole SMR result
eQTLGen
A list of all the result is available here:
datatable(eqtlgen, rownames = FALSE, filter="top", options = list(pageLength = 5, scrollX=T) )GTEX
A list of all the result is available here:
datatable(data, rownames = FALSE, filter="top", options = list(pageLength = 5, scrollX=T) )